접고 또 접고
- 노벨화학상과 단백질
- 글 재료공학부 2학년 김종흔
- 편집 항공우주공학과 1학년 임채민
서문
이번 2024년 노벨화학상은 AI를 빼놓고 얘기할 수 없습니다. 수상자 중에서 구글의 AI 연구원이 데미스 허사비스(구글 딥마인드 최고 경영자)와 존 M. 점퍼(구글 딥마인드 수석연구원), 이렇게 2명이나 된다는 점에서 말이죠. 또 다른 수상자인 데이비드 베이커(미국 워싱턴대 교수) 역시 그의 연구실 '베이커랩(BakerLab)'에서 단백질 설계 AI '로제타폴드'를 개발했습니다. 이들은 복잡한 단백질 구조를 밝히고, 더 나아가 새로운 구조의 단백질을 개발하는 데 AI를 접목한 공로를 인정받아 이번 상을 받을 수 있었습니다. 생명과학을 좋아하는 공학도로서 이번 노벨화학상에 두 번 놀랐습니다. X선 회절법으로 DNA를 발견한 지 겨우 70년 만에 2억 개 이상의 단백질 입체 구조를 모두 규명할 수 있음에 놀라고, AI 모델에 '진화'의 원리를 적용했다는 점에서 한 번 더 놀랐습니다. 이런 배경에서 그들이 만든 단백질 예측 AI인 알파폴드와 로제타폴드가 참 궁금해지는데요, 이번 기사에서는 공대생의 마음을 설레게 한 '알파폴드'와 '로제타폴드'를 함께 알아봅시다!

단백질 구조만 알면 돼!
우선 수많은 연구진이 단백질 입체 구조를 밝히는 데에 열중하는 이유부터 알아봅시다. 단백질은 생명 현상의 시작과 끝입니다. 생명체 내부의 화학반응을 매개하며 생명체 자체를 구성하기 때문이죠. 따라서 단백질을 정복하면 화학반응으로 대표되는 생명 현상 전반을 예측할 수 있고, 이를 통해 원하는 기능의 단백질을 설계할 수 있게 됩니다. 즉, 단백질 구조만 알 수 있다면 세상의 모든 병을 치료할 수 있다는 뜻이죠!
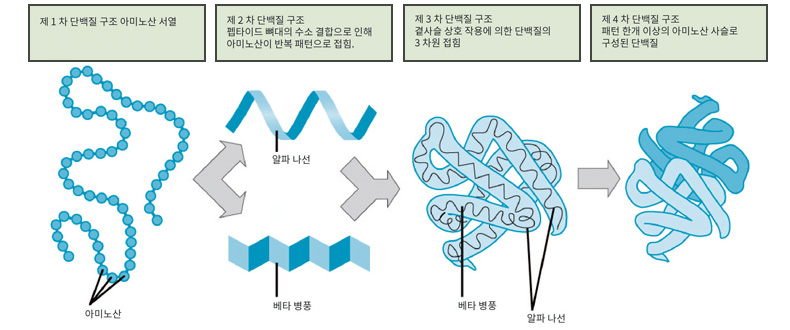
단백질은 흔히 아는 구부러진 물 분자 구조처럼 단순하지 않습니다. 단백질은 분자량이 30,000에서 50,000g/mol로 18g/mol인 물 분자와 비교했을 때 대략 2,000배 정도 무겁습니다. 거대한 단백질은 주변 아미노산 간의 인력과 척력의 상호작용으로 여러 번 접히고 꺾이죠. 게다가 자연에 존재하는 아미노산의 종류는 무려 20가지이고, 한 단백질에는 수백 개의 아미노산이 들어있습니다. 따라서 모든 경우의 수를 직접 계산하는 방식으로는 100년이 지나도 구조를 완전히 파악하기 어렵습니다.
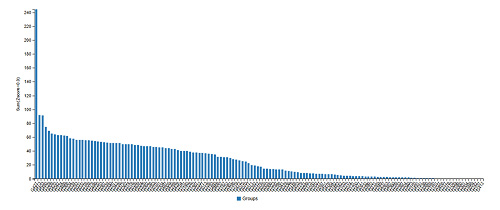
알파폴드가 이 문제를 '어떻게' 해결했는지 알아보기 전에, 얼마나 '잘' 해결했는지부터 알아봅시다. 그러기 위해선 CASP 14 대회 성과를 알아볼 필요가 있는데요. 2020년에 개최된 CASP 141)대회에서 알파폴드는 종합 평가를 의미하는 z-점수2)에서 244점으로 98점의 2위를 크게 압도했습니다. 또한, 단백질 구조 예측 정확도를 평가하는 GDT 점수가 90점 이상일 때 실험적으로 얻은 구조와 매우 유사하다고 간주하는데, 알파폴드는 평균 92.4점을 기록했습니다. 이는 알파폴드가 실제 연구에 도입되어도 무리가 없음을 보여줍니다.
알파폴드에 진화의 비밀이 담겨 있다고?

알파폴드 성능의 핵심인 다중서열정렬(MSA) 알고리즘을 이야기하려면 '진화(進化)' 얘기를 빼놓을 수 없습니다. 생명체의 진화는 염기 서열의 변화 그 자체입니다. 그리고 염기 서열의 변화는 아미노산 서열의 변화, 즉 단백질의 변화죠. 따라서 단백질 아미노산 서열 역시 진화의 법칙인 자연선택의 지배를 받습니다. 예를 들어 특정 아미노산 서열이 단백질의 기능에 중요하다면 조그만 변이에도 문제가 발생하는데요, 이는 개체의 생존에 치명적으로 작용하기 때문에 그런 변이를 가진 개체를 찾아보긴 어려울 것입니다. 반면에, 단백질 기능에 영향을 주지 않는 부위는 변이가 일어나도 개체의 생존에 문제가 없어 다양한 형태의 변이가 관찰될 것입니다.
이런 개념을 응용해 생명과학자들은 단백질의 아미노산 서열을 크게 '보존된 영역'과 '변이가 허용되는 영역'으로 나눴습니다. 이때 보존된 영역은 중요한 역할을 수행하며, 돌연변이가 거의 허용되지 않는 영역입니다. 주로 다른 물질과의 결합을 담당하거나, 단백질 구조 안정화를 담당하는 서열 부분이죠. 이들은 조금만 변형돼도 기능을 전부 잃거나 구조적으로 유지될 수 없기에 특정 서열들만 존재할 수 있습니다.
이제 보존된 영역에서 단백질 구조 안정화를 담당하는 서열들을 자세히 들여다보면 한 가지 현상이 관찰되는데요, 바로 '아미노산 공진화(共進化)'입니다. 이때 아미노산 공진화란 단백질의 구조적 안정에 기여하는 아미노산들이 진화 과정에서 쌍으로 변하는 현상을 의미합니다. 이런 아미노산 공진화의 특성을 이용하면, 앞서 언급한 MSA 알고리즘을 설계할 수 있는데요, 알고리즘의 작동을 더 자세히 이해하기 위해 특정 아미노산 서열의 입체 구조를 예측하는 상황을 가정해 봅시다. 우선 주어진 아미노산 서열과 비슷한 서열을 가진 단백질을 참고하면 좋겠죠? 그래서 MSA알고리즘에선 주어진 아미노산 서열과 유사한 단백질 구조들을 단백질 데이터베이스(PDB)에서 찾는 것부터 시작합니다. 이제 단백질의 입체 구조를 대략 설계할 차례인데요, 앞 단계에서 아미노산 공진화 쌍 정보를 확보했다면 우선 해당 아미노산 쌍이 서로 가까이 위치하도록 배치합니다. 공진화 쌍은 통계적으로 서로 가깝고 구조적으로 중요하기 때문이죠! 이렇게 중요 아미노산 쌍을 뼈대로 사용하면, 나머지 아미노산들을 뼈대에 알맞게 붙이는 건 식은 죽 먹기입니다.
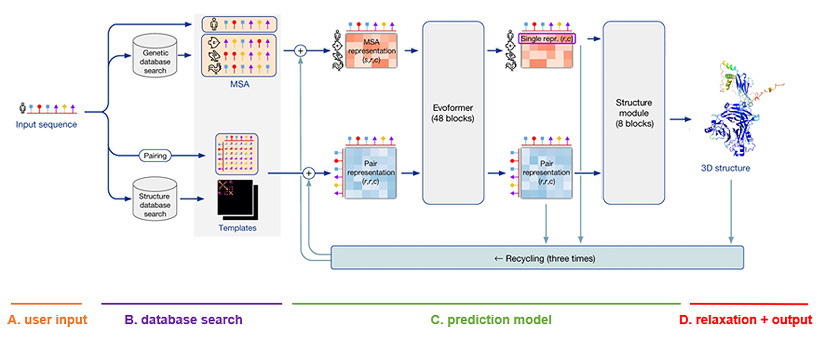
여기까지가 초기 구조를 만드는 과정입니다. 아직 MSA 알고리즘에서는 물리적으로 가능한 상황에 대해 학습하지 않았기에, 단백질 구조 데이터베이스에서 아미노산 간의 거리, 방향과 물리적 상호작용에 대한 정보를 학습합니다. 이는 앞선 초기 구조에 대한 정보와 Evoformer라 불리는 신경망 블록을 통해 단백질 구조가 더욱 정교해집니다. 끝으로 구조 모듈을 통해 아미노산의 3차원 좌표를 계산하는 과정을 거치면 구조 예측이 완료되는 것이죠.
생명과학자들은 AI 모델을 설계할 수 있도록 이론적 배경을 제공했을 뿐만 아니라 AI 개발에서 가장 중요한 '양질의 데이터'를 제공했습니다. 과학자들은 단백질을 끈질기게 연구해 왔고, 그 결과로 20만 개 이상의 단백질 구조와 서열에 대한 정보가 담긴 단백질 데이터베이스(PDB)가 탄생했습니다. 단백질 데이터베이스는 알파폴드의 학습 자료로 인공지능 모델의 디테일을 완성했죠. 그간 생명과학자들의 열정이 AI를 만나 꽃피운 셈입니다.
물론 몇 가지 숙제가 남아있는데요, 첫째로 단백질-단백질 상호작용이나 기타 이온과의 상호작용을 예측하지 못합니다. 특히 다른 물질과 형성한 결합 구조를 분석하는 데 부족한 성능을 보입니다. 둘째로 단백질의 동적인 변화 예측에도 약합니다. 단백질은 생명체 내부에서 온도, pH, 이온과 상호작용 등으로 끊임없는 변화를 수반하는 경우가 많은데, 알파폴드는 정적인 상태만 조사할 수 있어 활용할 수 있는 분야가 크게 제한되죠.
마지막으로 PDB에 정보가 부족한 단백질에 대해 예측 성능이 낮습니다. 앞서 언급한 MSA 알고리즘은 데이터베이스에 주어진 서열과 유사한 단백질들이 얼마나 많이 있는지에 따라 성능이 결정됩니다. 알파폴드의 목적은 잘 알려지지 않은 단백질 구조를 규명하는 것에 있으나, 역설적으로 단백질 구조에 대한 사전 정보가 부족하면 예측 신뢰도가 하락한다는 것은 치명적인 단점입니다. 이 단점은 자연에 존재하지 않는 아미노산이나 서열을 디자인하는 '데 노보(De novo) 단백질 디자인'에서 치명적으로 작용합니다.
데 노보(de novo) 단백질 디자인
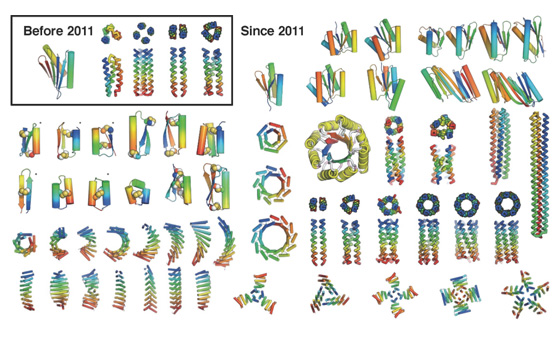
데 노보 단백질 디자인은 알려진 서열 정보만으로는 원하는 기능의 단백질을 구현하기 어려운 경우가 많아 등장했는데요, 아예 새로운 아미노산 서열을 설계하거나 자연에 존재하지 않는 아미노산을 이용해 기존에 없던 단백질을 설계합니다. 앞서 언급한 베이커 교수는 데 노보 단백질 디자인에 AI를 활용하기 위해 '로제타폴드(RosettaFold)'를 개발했습니다. 앞서 소개한 알파폴드는 실제 존재하는 단백질의 구조를 맞히고자 한다면, 로제타폴드는 원하는 기능을 수행할 단백질 구조를 설계하는 데에 초점이 맞춰져 있습니다. 따라서 단백질의 필요한 기능을 하기 위한 요소들을 결정하는 과정을 작동 첫 단계로 배치하는데요, 이 부분이 알파폴드와의 가장 큰 차이점입니다. 물론 이후의 과정은 거의 비슷합니다. 단백질의 구조를 안정화하는 아미노산 골격을 설계하고, 이후엔 초기에 형성된 구조가 더 안정적인 구조가 되도록 서열을 최적화하며 마무리하죠.
이렇게 합성된 단백질이 원하는 기능을 수행하고 구조적으로 안정하다는 것까지 검증됐다면, 산업적인 대량생산까지 이어지는 것이 로제타폴드의 최종 목표입니다. 그런데 자연에 없는 단백질은 어떻게 대량생산할 수 있을까요?
과학자들은 단백질이 DNA의 정보로부터 합성됨에 주목했습니다. 단백질의 아미노산 순서에 대한 정보는 DNA에 모두 저장돼 있으니까, DNA를 수정하면 단백질 순서도 바꿀 수 있다는 발상입니다. 이를 바탕으로 비슷한 단백질을 생산하는 박테리아의 DNA를 조작해 원하는 단백질 합성을 유도하면 새로운 단백질을 합성하는 세포 공장을 만들 수 있겠죠!
로제타에서 단백질을 개발하고 실제로 대량생산까지 이어지는 과정에는 아직 극복해야 할 문제들이 많습니다. 첫째로 로제타는 이제껏 없었던 단백질을 만드는 만큼, 알파폴드보다도 정확도가 떨어집니다. 게다가 예측한 단백질이 지금껏 없었던 물질이라 검증하는 과정도 까다롭죠. 둘째로 자연적에 존재하지 않는 아미노산을 이용해 단백질을 합성하기 위해선 박테리아의 DNA 서열을 바꾸는 것뿐만 아니라 기존 단백질 합성 시스템 자체를 수정해야 합니다. 이는 세포 소기관 단위부터의 제어를 의미하는데, 이는 굉장한 도전입니다.
생명공학은 그간의 노력과 함께 강력한 도구를 손에 얻었습니다. 이제 과학자들은 자연에서 발견하는 것이 아니라, 원하는 기능을 가진 단백질을 디자인하는 새로운 패러다임으로 전환하고 있습니다. 새로운 생명공학과 함께 펼쳐질 세상을 기대해 봅시다.
참고
- 1) CASP14: 단백질 구조 예측 AI의 발전을 평가하는 국제 경진대회.
- 2) z-점수: 특정 예측 모델의 성능이 평균에서 얼마나 벗어났는지를 표준편차 단위로 나타낸 값.
참고 논문
- Lovelock, S.L., Crawshaw, R., Basler, S. et al. The road to fully programmable protein catalysis. Nature 606, 49-58 (2022).
- Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583-589 (2021).
- Minkyung Baek et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871-876 (2021).
- Anishchenko, I., Pellock, S.J., Chidyausiku, T.M. et al. De novo protein design by deep network hallucination. Nature 600, 547-552 (2021).
그림 출처
- 그림1. https://www.france24.com/en/live-news/20241009-%F0%9F%94%B4-david-baker-demis-hassabis-john-jumper-win-nobel-chemistry-prize-for-work-on-proteins
- 그림2. https://www.labxchange.org/library/items/lb:LabXchange:510e7f5c:html:1
- 그림3. https://predictioncenter.org/casp14/zscores_final.cgi
- 그림4. https://www.ipd.uw.edu/2017/01/big-data-shapes-the-fold-for-of-hundreds-of-protein-families/
- 그림5. https://elearning.vib.be/courses/alphafold/lessons/the-alphafold-pipeline/topic/overview-of-the-architecture/
- 그림6. https://www.bakerlab.org/2016/09/16/the-coming-of-age-of-de-novo-protein-design/